For programmers new to ObjectScript, one question will inevitably arise: “What is the difference between methods and class methods?” A typical answer would be: “A class method applies to a class, but a method applies to an instance of that class.” While that answer is correct, it lacks important information on how these methods differ, and how they are used in ObjectScript. Many things could be written as either. For instance, suppose we had a class called “User.Person” with a property called “Name”. If we wanted to create a method to write that name, we could do either of the following:
Method WriteName()
{
write ..Name
}ClassMethod ClassWriteName(id)
{
write ##class(User.Person).%OpenId(id).Name
}In many cases, it does not matter what we choose. However, there are some compelling reasons why we might prefer one over the other. We will dive deeper into this topic today to find that out.
Methods: General Guidelines
Methods are sometimes referred to as instance methods for the sake of clarity. They are selected when the method does something to or with a specific instance. Methods are the tools to use when an in-memory instance of an object is required. This, of course, means that they should only be defined in object classes. Since they are operating on an instance, they can refer to that object’s properties using the double-dot syntax, as in the WriteName method above. One of the most basic examples we all quickly become familiar with is %Save(). If we do not have an instance of the object, it is impossible to know what to save.
Instance methods are also an excellent choice when a method must be able to operate on an instance of an object before that instance is saved (or inserted via SQL if the class extends %Persistent). %ValidateObject is a great example of this situation. We need to make sure the object is valid before we save it.
Class Methods: General Guidelines
Although class methods are defined within a class, they are not tied to a specific instance of a class. If you are familiar with some other programming languages, you may be accustomed to calling such methods “static methods”. Since these methods are not called on a specific instance of the class, you cannot employ the double-dot syntax to refer to properties of the class. You must get an instance of an object instead, and then use the dot syntax to refer to that instance’s properties.
Class methods must be used when a method does not have access to an instance. Methods that create a new instance of an object, a.k.a. “constructors”, in most cases, will be class methods. One situational exception to this is %ConstructClone. It builds a new instance of an object copied from an existing instance, so it is defined as an instance method. The default constructor for most objects is %New(), but you may also want to write some of your own.
Class methods are favored when a method operates on multiple instances of a class. We could transform such methods into instance methods, and make them work just fine, but doing it would not make much sense. For example, if we wanted to convert all of our names to uppercase, we could use the following method:
ClassMethod AllToUpper(){
set stmt = ##class(%SQL.Statement).%New()
set query = "SELECT ID FROM SQLUser.Person"
do stmt.%Prepare(query)
set result = stmt.%Execute()
while result.%Next(){
set myPerson = ##class(User.Person).%OpenId(result.GetData(1))
set myPerson.Name = $ZCONVERT(myPerson.Name,"U")
do myperson.%Save()
}
}If we defined this method as an instance method, it would still do the same thing. However, we do not usually do that because it would require us to take the extra step of either creating or opening an instance of the class before calling the method, which would be a waste of time and resources.
Likewise, class methods are picked when a method operates on no instances of an object. Suppose we have a class parameter defined, in this case, to override the caption parameter, do the following:
/// Optional name used by the <i>Form Wizard</i> for a class when generating forms.
Parameter CAPTION = "People";Since parameters are shared among all members of a class (a “static variable” as called by some other programming languages), it is not necessary to access a specific instance of a class to refer to that parameter. If we wanted to write that parameter’s value, we could do it as shown below:
ClassMethod WriteCaption()
{
write ..#CAPTION
}Once again, in this case, we could convert it into an instance method. However, it would be just as inefficient as in our previous example.
Class Methods: Special Cases
So far, these all have been general programming guidelines, which, by the way, you could break and still get the desired result. However, there are a few specific cases when you must use a class method.
If you want to call a method in a trigger, it must be a class method. Triggers can be powerful, but they also have some extra rules and special syntax since they are executed when data is in flux. They allow you to specify whether you want to refer to the old value or the new value of a changing property, and that is precisely why they cannot use the double-dot syntax. Suppose I want to log the UTC timestamp of a record being deleted from my Person. I could define the following method and trigger to accomplish that as indicated below:
ClassMethod LogDelete(id)
{
set ^LogDelete("Person",id) = $ZDATETIME($ZTIMESTAMP)
}
Trigger LogDelete [ Event = DELETE, Foreach = row/object ]
{
do ##class(User.Person).LogDelete({%%ID})
}Then, when I delete a record from that table, I will see a global set like the following one:
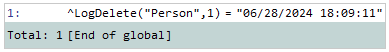
If we want a method to be exposed as an SQL procedure, it must be a class method. If my name field is just a first and last names separated by a space, “David Hockenbroch,” but we want to have a stored procedure to return it in the way favored by Elon Musk when it comes to his phone order, “Hockenbroch, David,” we can write a simple function like the one below:
ClassMethod AlphaName(id) As %String [ SqlProc ]
{
set myPerson = ##class(User.Person).%OpenId(id)
return $P(myPerson.Name," ",2)_", "_$P(myPerson.Name," ",1)
}Note the SqlProc keyword in the function definition. This keyword will tell the compiler to project this method as an SQL procedure called Person_AlphaName. We can then exploit it in a query as follows:
select name, person_alphaname(id) As "AlphaName" from person
We will see the next result:
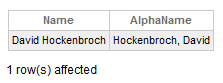
Finally, I can utilize a class method to define how an SQL computed property is calculated. Suppose I have the following property in my class:
Property AlphaName As %String [ SqlComputed ];When we employ the SqlComputed keyword, we must define the computation to be operated. One way to do that is with the SqlComputeCode keyword. The other one is to specify a class method that returns the desired value. This method must have the same name as its property followed by “Computation.” Consider the following example:
ClassMethod AlphaNameComputation(cols As %PropertyHelper) As %String
{
return $P(cols.getfield("Name")," ",2)_", "_$P(cols.getfield("Name")," ",1)
}If we do that, then the property will be calculated any time the object is inserted or updated. Note the %PropertyHelper argument that gets passed to this method. This object allows you to access the properties of the individual instance whose property is being calculated despite this being a class method. You can use the getfield property of that object and give it a property name to get that property. In this case, we just have a column, so we should use the SQL:
select Name, AlphaName from person
The result you get then will be identical to the one before:
In Conclusion
I hope this review of methods was detailed enough and included everything you need to know to determine which kind of method to use. If I have missed anything, please, let me know in the comments below!